
| |
Manual |
|
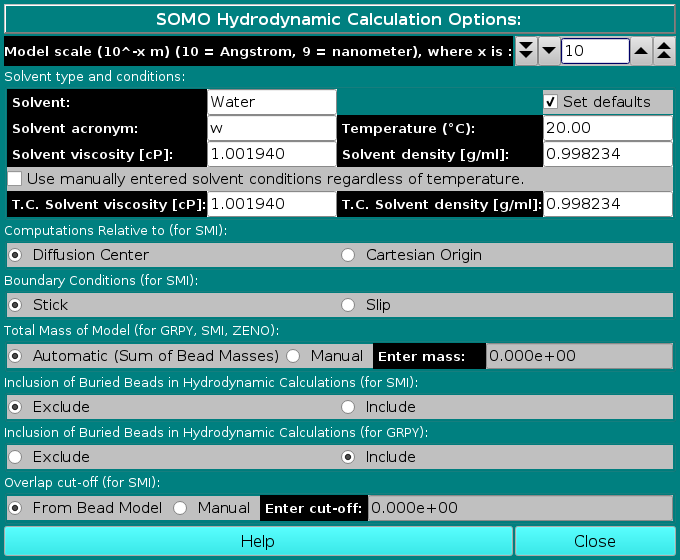
In this module, various options controlling the computations of the hydrodynamic parameters for bead models can be set. The Model units (-10 = Angstrom, -9 = nanometer) field sets the scale units of the bead model being examined. Models derived from PDB structures are in Angstrom units, but the user might wish to compute the hydrodynamic parameters for bead models coming from different sources, for instance from small angle solution scattering data, which might be in other units like nanometers. The field sets the exponential relating the units to meters (default: -10). The Solvent type and conditions: box allows the definition of the solvent conditions to which the hydrodynamic computations are referred. The default conditions (Water @ 20 °C) can be automatically set by selecting the Set defaults checkbox. Otherwise, enter a text description of the solvent in the Solvent field (default: Water) and its acronym (max 3 characters; default: w) in the Solvent acronym field. The temperature (°C) can be set in the Temperature (°C): field (default: 20 °C), which will also affect the value of the partial specific volume vbar, according to the equation: vbar(T °C) = vbar(ent) + [4.25 x 10-4 (T - T(ent))]
where vbar(ent) is either the vbar value calculated by the program at T(ent)=20 °C, or the manually entered vbar with its associated T(ent). See here for how to control the vbar parameter.
To allow for more freedom in setting the solvent conditions, from the February 2021 release a new option is present: by selecting the checkbox "Use manually entered solvent conditions regardless of temperature", the values present in the "T.C. Solvent viscosity [cP]:" (default: 1.001940) and "T.C. Solvent density [g/ml]:" (default: 0.998234) will be used (T.C. stands for "Temperature Corrected", meaning that these values have been measured or calculated at a specific temperature). Note that these settings will be used by all the available computational methods, SMI, ZENO, and GRPY . The other options will affect either all or just some of the available computational methods. The "Computations Relative to (for SMI):" box presents two alternative options that will be applied only to the SMI method, Diffusion Center and Cartesian Origin. The differences between the two are subtle, and are fully described in Garcia de la Torre and Bloomfield, Q. Rev. Biophys. 14:81-139, 1981 (default: Diffusion Center). The "Boundary Conditions (for SMI):" box also has two alternative options, valid only for SMI, Stick (6πη0) or Slip (4πη0). For structures whose size is greater than the hypothetical solvent molecules, the stick boundary conditions apply. For the SMI method, the slip boundary conditions should perhaps be used only when the size of the entire molecule is comparable with the size of the solvent molecules (see Venable and Pastor, Biopolymers 27:1001-1014, 1988; Garcia de la Torre and Bloomfield, Q. Rev. Biophys. 14:81-139, 1981) (default: stick).
The "Total Mass of Model (for GRPY, SMI, ZENO):" box, that applies to all methods, has two checkboxes allowing the user to select or override the Automatic (Sum of Bead Masses) computation of the mass of the model obtained by summing over the mass assigned to each bead. This can be done by selecting the Manual checkbox and entering a value in the Enter mass field. For normal operations on models generated by US-SOMO from PDB files, the Automatic option will do fine, except if a relevant number of non-coded or incomplete residues are skipped or are modeled with the Automatic Bead Builder (because then the total mass will be appreciably underestimated). If a Manual value is entered
in this field (and the Automatic (Sum of Bead Masses) checkbox is deselected), a message will be displayed in the progress window ("ATTENTION: MW = ....."). This should avoid the use of an incorrect external total mass value resulting by inadvertently leaving the Manual option selected from a previous model-generating session.
The "Inclusion of Buried Beads in Hydrodynamic Calculations (for SMI)" box, valid only for the SMI method, allows to either Exclude or Include the beads labeled as buried in the hydrodynamic computations by SMI for SoMo models without overlaps. We have demonstrated (Rai et al., Structure 13:723-734, 2005; Brookes et al., Eur. Biophys. J., 39:423-435, 2010) that for these models excluding the buried beads has no effect on the translational diffusion properties. Moreover, as the memory required by the full supermatrix inversion procedures implemented in the SMI hydrodynamics computations module grows exponentially with the number of beads employed, excluding the buried beads from the computations allows for both a faster processing and the capability of processing bigger structures (default: Exclude). The "Inclusion of Buried Beads in Hydrodynamic Calculations (for GRPY)" box, valid only for the GRPY method, allows to either Exclude or Include the beads labeled as buried in the hydrodynamic computations by GRPY. As GRPY calculates the rotational diffusion coefficients and the intrinsic viscosity with a sound procedure, the effect of excluding a relevant portion of the beads, especially for atomic-sized beads such as in the vdW models, needs an in depth investigation. As of February 2021, preliminary tests indicate that reducing by ∼50% the number of the used beads by using an ASA probe radius of 1 Å and a cut-off of 1% of the bead theoretical surface area (see here) has a ∼+0.1% effect on the translational diffusion coefficient, a ∼-0.13% on the harmonic mean of the rotational relaxation time, and a ∼-0.22% on the intrinsic viscosity values for vdW models of lysozyme. While excluding buries beads from the computations surely allows for both a faster processing and the capability of processing bigger structures by the GRPY method, for the moment being the default option is to include the buried beads. Finally, the "Overlap cut-off (for SMI): box, valid only for the SMI method, allows the user to select a different bead overlap cut-off when checking the model before the hydrodynamic computations (it should be recalled that the hydrodynamic interaction tensor used by the SMI method is valid only for non-overlapping beads). If an overlap exceeding the threshold is found between any couple of beads, the computations are halted. An overlap cut-off is already present in the Bead Overlap Reduction set-up options, and if the bead model has been internally generated, then the From Bead Model checkbox should be selected. For models generated in other ways, the tolerance can be increased (at your own risk!) by selecting the Manual checkbox and entering a value in the Enter cut-off field. If this cut-off is different from the one present in the Bead Overlap Reduction module, a message alerting of the different cut-off value will appear in the progress window when the SMI hydrodynamic computations are started (default: From Bead Model). www contact: Emre Brookes
This document is part of the UltraScan Software Documentation
distribution. Last modified on December 18, 2020. |